Das Wichtigste
Der Morbus Wilson, auch als Wilson-Krankheit oder hepatolentikuläre Degeneration bezeichnet, ist eine genetisch bedingte Kupferspeicherkrankheit. Durch eine zunehmende Funktionsstörung vor allem der Leber und des Gehirns verläuft sie unbehandelt tödlich. Die Krankheit trägt den Namen des Neurologen Kinnier Wilson, der sie 1912 erstmals beschrieben hat.
Komplikationen: Zu den wichtigsten Auswirkungen der Kupferspeicherung zählen folgende:
- eine fortschreitende Vernarbung der Leber (Leberzirrhose),
- eine toxische Hirnschädigung mit erheblichen Störungen bei Bewegungsabläufen und mit Persönlichkeitsveränderungen,
- die „Wilson-Krise“ mit akutem Leberzerfall und Blutzersetzung (Hämolyse),
- die Einbeziehung von Nieren, Herz und Skelett, was zu einer Vielfalt weiterer Symptome führt.
Symptome: Die ersten Symptome betreffen die einer Leberzirrhose mit einer Lebervergrößerung und einem Nachlassen aller Leberleistungen (Leberinsuffizienz), einer Ösophagusvarizenblutung, Leberhautzeichen und Gelbsucht. Neurologische Störungen mit Bewegungsstörungen und Schluckstörungen werden in der Regel erst dann auffällig, wenn die Leber beginnt, insuffizient zu werden. Dann beginnt auch das Gehirn Kupfer einzulagern. Es entwickeln sich verschiedene Auffälligkeiten, wie Ataxie, Tremor und Störungen des Gangbilds. Solche Symptome können bereits im Kindesalter auftreten.
Diagnostik: Die Diagnose beruht auf dem Nachweis von Kupfer und Coeruloplasmin im Blut und einer erhöhten Kupferausscheidung mit dem Urin (24-Stunden-Urin), die durch das Basismedikament D-Penicillamin erheblich verstärkt wird. Eine genetische Analyse bestätigt die Diagnose und dient auch als familiäres Screening.
Therapie: Die Behandlung zielt auf eine Verminderung des Kupferspeichers im Körper. Dies kann erreicht werden durch
- Vermeidung einer Zufuhr von Kupfer über die Nahrung und über Flüssigkeiten (Vorsicht: Kupferleitungen, Kupferkessel; diätetische Beratung erforderlich),
- einer Aufnahme von Kupfer über den Darm durch Zink-Präparate und
- eine vermehrte Ausschwemmung über die Nieren durch Kupfer-bindende Komplexbildner (Chelatbildner), wie D-Penicillamin und Trientin.
Perspektive: Eine Stammzelltherapie ist in Entwicklung / Erprobung; sie kann den Kupferstoffwechsel dauerhaft korrigieren.
→ Kupferstoffwechsel
→ Wilson-Krise
→ Therapie des Morbus Wilson
Epidemiologie
Die Häufigkeit der Wilson-Krankheit liegt bei ca. 1-3 pro 100.000 Einwohner. Männer sind geringfügig häufiger betroffen als Frauen. 1 Neuere Bewertungen ergaben eine höhere Prävalenz von etwa 1 pro 20.000 2 bzw. von 12,7 pro 100.000. 3
Ursache
Der Wilson-Krankheit ist eine monogenetische, autosomal-rezessiv vererbte Speicherkrankheit, die zur Kupferüberladung und damit zu einer toxischen Schädigung verschiedener Organe des Körpers führt.
Genetische Grundlage
Verursacht wird der Morbus Wilson durch Mutationen im ATP7B-Gen auf Chromosom 13. Die geschätzte Trägerrate des abnormen Gens beträgt 1 pro 90 4. Das Genprodukt ist eine spezielle ATPase (ATP7B). Sie ist für den Kupfertransport in der Leber und die Ausscheidung in die Galle verantwortlich. Inzwischen sind über 250 Mutationen bekannt, die häufigste ist (in etwa 15 % der homozygoten Fälle) H1069Q. Die Vielfalt der Mutationen erklärt die unterschiedlichen Ausprägungen der Krankheit.
In seltenen Fällen wurde ein Morbus Wilson beschrieben, bei dem keine Mutation des ATP7B-Gens nachweisbar war; die Ursache blieb unbekannt 5. Inzwischen sind über 100 verschiedene Mutationen bekannt geworden 6, und es werden immer wieder neue entdeckt 7.
Auswirkungen sind in jedem Fall ein verringerter Einbau von Kupfer in Apoceruloplasmin und eine verringerte Ausscheidung von Kupfer mit der Galle. 8 9
Mencke´sche Erkrankung
Die Mencke´sche Erkrankung ist mit dem Morbus Wilson verwandt und durch einen Defekt des Gens ATP7A gekennzeichnet, das – wie das Wilson-Gen (ATP7B) – für eine Kupfer-transportierende ATPase kodiert, die Kupfer aus Körperzellen heraustransportiert. Es handelt sich um eine fortschreitende Neurodegeneration und Bindegewebsstörungen. Typisch ist „krauses” Haar. Eigenartigerweise ist der Kupfergehalt der Leber und des Gehirns niedrig, während der anderer Gewebe hoch ist. Bei den meisten Patienten treten im Alter von etwa 2 bis 3 Monaten therapieresistente Anfälle auf. Nach wenigen Monaten werden Entwicklungsrückschritte auffällig. Die Kinder überleben meistens keine 3 Jahre. 10
Entstehung
Die Kupfer-Speicherung beginnt in der Leber. Je überladener die Leber ist, desto mehr weitere Organe werden in die Kupferaufnahme einbezogen. So findet sich ein krankheitstypischer brauner Farbring am Auge (Kayser-Fleischer-Kornealring, s. u.) erst dann, wenn nach der Leber auch das Gehirn bzw. zentrale Nervensystem in die Kupferspeicherung einbezogen ist. 11 12
Cuproptose: Die Cuproptose ist eine besondere Art des organisierten Zelltods, die durch eine übermäßige intrazelluläre Konzentration von Kupfer in Zellen verursacht wird. Der Mechanismus hat Aufmerksamkeit erlangt, da er zur Bekämpfung von Krebs eingesetzt werden könnte. 13 Es wird vermutet, dass er dafür verantwortlich ist, dass Wilson-Patienten mit Leberzirrhose weniger häufig Leberkrebs bekommen, als Patienten mit Zirrhose anderer Ursache.
Kupferansammlung in Leberzellen: Ursache der Kupferansammlung in den Leberzellen (Hepatozyten) ist ein defekter Kupfertransport innerhalb der Zellen. Damit Kupfer in die Galle ausgeschieden werden kann, muss es zum Trans-Golgi-Netzwerk transportiert werden.
- Die dafür notwendige Energie liefert eine ATPase, welche das Kupfer dem Transportmolekül Coeruloplasmin übergibt. Erst mit diesem zusammen kann es in die Galle ausgeschieden werden. Der Wilson-Krankheit liegt ein Defekt dieser ATPase vor.
Es sammelt sich zunächst in der Leber und später im gesamten Körper an. Durch oxidativen Stress kommt es zu einer Schädigung der Mitochondrien und damit der Leberzellen, die defekt werden und zugrunde gehen. Wenn viele Leberzellen auf einen Schlag zugrunde gehen, entwickelt sich durch die große Menge des frei werdenden Kupfers eine lebensbedrohliche Wilson-Krise mit hohen Kupferspieleln im Blut. Durch ihn kommt es zur für die Wilson-Krise typischen Auflösung roter Blutzellen (Hämolyse).
Störung der Bildung von Coeruloplasmin: Unabhängig vom Kupferexkretionsdefekt besteht beim Morbus Wilson eine Störung der Bildung von Coeruloplasmin, was zu einer verminderten Coeruloplasminkonzentration im Blut führt. Dies wiederum ist für die übermäßige Kupferaufnahme und die Schädigung verschiedener Organe von Bedeutung.
Symptomatik und klinisches Bild
Primär hepatische Symptomatik
Bei der Erstdiagnose eines Morbus Wilson finden sich i. d. R. eine Leber- und Milzvergrößerung (Hepatosplenomegalie) und in über 40 % Ösophagusvarizenblutung, Ikterus, Leberhautzeichen.
Primär extrahepatische Symptomatik
Zur Diagnose führen häufig nicht Lebersymptome, da die Kupferspeicherung in der Leber lange Zeit keine besonderen subjektiven Beschwerden hervorruft, sondern Symptome, die von geschädigten Körperorganen ausgehen.
Augen
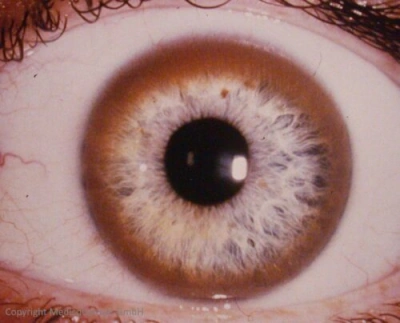
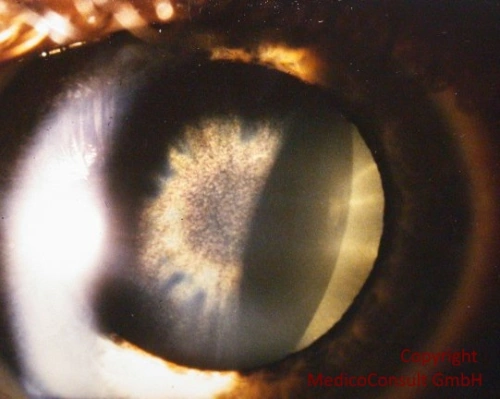
Sonnenblumenkatarakt bei Mobus WilsonAugensymptome sind Hinweise auf eine neurologische Beteiligung der Kupferspeicherkrankheit:
- Kayser-Fleischer-Kornealring: Er ist Folge einer Kupferablagerung und in ca. 80% der klinisch manifesten Fälle nachweisbar. Er stellt kein Frühsymptom dar und kommt in der Regel nur bei gleichzeitigem Vorliegen neurologischer Symptome vor. Sein Nachweis ist ein wichtiges klinisch-diagnostisches Kriterium für den M. Wilson. Unter der Therapie ist er reversibel. Seine Kontrolle dient daher der Therapiekontrolle.
- Sonnenblumenkatarakt: Sie ist nicht so häufig, beeinträchtigt die Sehkraft kaum, tritt meist mit dem Kayser-Fleischer-Kornealring gemeinsam auf und ist unter der Therapie ebenfalls reversibel.
Zentralnervensystem
Symptome einer Mitbeteiligung des zentralen Nervensystems 14 15 finden sich bei der Erstmanifestation bereits in 34% der Fälle. Zu ihnen gehören:
- Störungen der Motorik
– Dystonie: Fehlhaltungen, Verkrampfungen, Grimassieren,
– Koordinationsstörungen (Fein-, später Grobmotorik),
– gestörtes Gangbild (z. B. stolpernd), - Sprechstörungen (Dysarthrie),
- Tremor (Intentions-, später Ruhetremor),
- pathologische Reflexe,
- Dysphagie,
- Persönlichkeitsstörungen und Verhaltensauffälligkeiten (bei der Erstmanifestation in 10 %).
In einer Zusammenstellung von 69 Fällen (Alter 13,6 +/- 6,6 Jahre) fanden sich folgende neurologische Symptome: Tremor (43,6 %), Dystonie (41,8 %), Parkinsonismus (52,1 %), Chorea (10,1 %), Ataxie (1,4 %). Das Putamen, der Nucleus caudatus und der Thalamus waren am häufigsten betroffen, danach auch Mittelhirn, Kasel und Pons. 16
Das Vorliegen einer neurologischen Symptomatik verschlechtert die Prognose einer Wilson-Krankheit. 14
Nieren
Eine Einschränkung der Nierenfunktion (Niereninsuffizienz) ist eine Spätmanifestation des Morbus Wilson. Es kann sich eine Nephrokalzinose und Nephrolithiasis (durch Störung der Kalziumausscheidung) entwickeln. Die Neigung zu Nierensteinen wird durch eine renal-tubuläre Azidose bei einer veränderten der Säure-Basen-Ausscheidung erklärt. 17
Knochen, Skelett
Skelettsymptome gehören ebenfalls zur Spätmanifestation und sind durch Knochenverdünnung (Osteomalazie oder Osteoporose) und Einbeziehung der Gelenke (Osteoarthritis durch Kupferablagerungen) gekennzeichnet. 18 Die Osteomalazie ist renal bedingt und wird durch eine Einschränkung der Phosphat- und Bikarbonatresorption erklärt. Sie stellt eine metabolische Knochenkrankheit dar. 19 Da erst Spätstadien der Wilson-Krankheit von dieser Komplikation betroffen sind (mit neurologischen Ausfällen), wird eine Routineüberprüfung des Knochenstatus bei der Wilson-Krankheit nicht für erforderlich gehalten. 20
Blut
Im Rahmen einer akuten Wilson-Krise als Folge eines akuten Leberversagens kommt es zu einer akuten Hämolyse (Zerfall der roten Blutkörperchen, bei der Erstmanifestation in 10 – 15 %), Leukopenie, Thrombopenie und hämorrhagischer Diathese (Blutungsneigung).
Herz
Bei fortgeschrittener Erkrankung kommt es durch Kupfereinlagerungen zu einer Kardiomyopathie mit Rhythmusstörungen und Neigung zur Hypotonie.
Diagnostik des Morbus Wilson
Die Wilson-Krankheit beginnt in aller Regel weit vor den 40-sten Lebensjahr symptomatisch zu werden. Dennoch wird die Krankheit in seltenen Fällen auch später noch diagnostiziert. Unter den Differentialdiagnosen einer chronischen Leberkrankheit, die im Alter von über 40 J erstmals diagnostiziert wird, spielt der Morbus Wilson daher kaum eine Rolle. Allerdings sollte auch dann noch an ihn gedacht werden, da eine Leberzirrhose lange Zeit übersehen werden kann, und da auch vielfach neurologische Symptome lange Zeit fehlinterpretiert werden. 21
Die Diagnostik stützt sich wesentlich auf Parameter des Kupferstoffwechsels im Blut (s. u.). Wegen der Vielfalt der Mutationen des Wilson-Gens ist eine molekulargenetische Untersuchung aufwendig.
Laboruntersuchungen
Diagnostik der Wilson-Krankheit
Folgende Befunde sind zur Diagnosestellung und zur Verlaufskontrolle erforderlich:
- Coeruloplasmin (in fast 90 % erniedrigt; Cut-off z.B. bei 150 mg/l)
- Kupfer i. S.: erniedrigt (in ca. 90 %),
– freies Kupfer im Serum: erhöht (praktisch immer über 50 µg/dl),
– Kupfer im 24-Stundenurin: über 100 µg/Tag erhöht; besonders starke Kupferausscheidung nach Gabe von Chelatbildnern, z.B. Metalcaptase.
Coeruloplasmin: Das wichtigste Kriterium für den Beginn der Diagnostik ist Coeruloplasmin. Werte von < 200 mg/l gelten als stark hinweisend. Sie sind in 85 % bis 90 % erniedrigt. Laut einem Cochrane-Review erreichte ein Cut-off von 0,2 g/l (nach Leipziger Kriterien) eine Sensitivität von 77,1 % bis 99 % und eine Spezifität von 55,9 % bis 82,8 %. Bei einem Grenzwert von 0,1 g/l stieg die Spezifität auf 96,6 % bis 100 %, wobei die Sensitivität auf 78,9 % sank. 22 In einer chinesischen Arbeit wird ein Cutoff von 150 mg/l empfohlen. 23
Kupferausscheidung im 24h-Urin: Bei einem Grenzwert von 0,64 bis 1,6 μmol/24 Stunden (nach Leipzig-Kriterien) fand sich eine Sensitivität von 50,0 % bis 80,0 % bei einer Spezifität von 75,6 % bis 98,3 %. 22
Die Molekulargenetik: Eine Bestimmung des Wilson-Gens (ATP7B-Gen, s. o.) sichert die Diagnose. Sie sollte im Fall eines Mutationsnachweises auch bei engen Blutsverwandten durchgeführt werden. Inzwischen sind über 500 Missense-Mutationen bekannt. 24 25
→ Laborwerte bei Leberkrankheiten
Diagnostik von Folgeschäden: Die wichtigsten Folgeschäden, die über Laborwerte zu kontrollieren sind, sind Leberinsuffizienz, Niereninsuffizienz und Hämolyse.
- Leberwerte inkl. Syntheseleistungsparameter der Leber: Sie sollen über eine mögliche Leberinsuffizienz Auskunft geben, wie sie in späten Stadien der Krankheit auftreten kann. Die alkalische Phosphatase kann ungewöhnlicherweise im Bereich der unteren Normgrenze oder erniedrigt ausfallen.
- Nierenwerte (ggf. inkl. 2-Stunden-Clearance): sie sollen eine mögliche Niereninsuffizienz anzeigen (Spätsymptom).
- Blutbild und Hämolysezeichen: durch krisenhaft aus den geschädigten Leberzellen frei werdendes Kupfer kann es zu einer Hämolyse kommen. In diesem Fall sind freies Hämoglobin, niedriges freies Haptoglobin und erhöhtes indirektes Bilirubin zu gewärtigen, sowie durch rasche Nachbildung roter Blutkörperchen eine Retikulozytose.
Bestimmung von Kupfer in einer Gewebeprobe
Histologischer Nachweis von Kupfer mit der Rhodaninfärbung (nicht Rhodamin!) und von Kupfer-Proteinkomplex mit Orcein. Sie können in frühen Stadien negativ sein. Am zuverlässigsten gibt die Atomabsorptionsspektrophotometrie die Kupferkonzentration im Lebergewebe an.
Kupferwerte bei der Wilsonkrankheit: atomspektrometrisch 250-3.000 µg/g Trockengewicht bei Homozygoten, 55 – 250 µg/g Trockengewicht bei Heterozygoten. Wegen ungleichmäßiger Verteilung des Kupfers ist ein „sampling error“ möglich. Ein erhöhter Kupfergehalt der Leber kommt differenzialdiagnostisch auch bei chronischen Cholestasen, z. B. PBC und PSC, und bei angeborenen Gallengangsdefekten, wie dem Alagille-Syndrom, vor.
Nachweis einer Hirnschädigung
Eine Schädigung der Basalganglien durch Kupferablagerungen lässt sich durch bildgebende Verfahren (Neuroimaging, z. B. durch MRT) relativ sicher nachweisen.
Ablauf der Diagnostik
Erste Hinweise auf einen Morbus Wilson können folgende sein:
- eine ungeklärte Leberzirrhose,
- eine ungeklärte neurologische Symptomatik oder
- ein akutes Leberversagen mit hämolytischer Krise im Alter zwischen 6 und 40 Jahren.
Wenn an die Möglichkeit eines Morbus Wilson gedacht wird, folgt eine Bestimmung von Kupfer im Serum (gesamtes und freies Kupfer) und im 24-Stunden-Urin, ergänzt durch die Bestimmung von Coeruloplasmin.
Durch niedriges Coeruloplasmin und hohe Kupferausscheidung (im 24-Stundenurin) sowie durch den Nachweis von Läsionen der Basalganglien durch Neuroimaging kann die Diagnose eines Morbus Wilson bereits sehr wahrscheinlich gemacht werden.
Durch eine Leberbiopsie mit Kupferbestimmung im Lebergewebe kann die Diagnose gesichert und gleichzeitig die Ausprägung des Leberschadens abgeschätzt werden.
Eine genetische Sicherung ist nicht immer erforderlich, kann aber zur Diagnostik von blutsverwandten Genträgern sinnvoll sein.
Es folgen Untersuchungen bezüglich Folgeschäden an Gehirn, Augen, Nieren, Bewegungsapparat (Knochen, Gelenke), Herz, Blut.
Differentialdiagnosen bei hochakutem Beginn (Wilson-Krise)
Hinweise auf einen Morbus Wilson bei akutem Beginn sind anamnestische Angaben von frühkindlichen Lebererkrankungen oder neurologischen Erkrankungen, Nachweis einer nichtsphärozytären, Coombs-neg. hämolytischen Anämie, Leberversagen mit auffallend geringer Aktivität der Transaminasen und auffallend verminderter Aktivität der alkalischen Phosphatase.
Bei einer Wilson-Krise können eine fulminante Hepatitis oder andere Ursachen einer akuten Leberdystrophie schwierige Differenzialdiagnosen darstellen, insbesondere, wenn die Kupferspeicherkrankheit bis dahin noch unbekannt ist. Zu Differenzierung können helfen:
- Leberwerte erhöht und zudem Zeichen von Blutzerfall (Hämolyse),
- Kupfer i. S. (ist bei der Wilson-Krise meist stark erhöht),
- Kayser-Fleischer-Kornealring fehlt meist noch bei der Erstmanifestation,
- Coeruloplasminkonzentration (bei 10 % – 15 % der an M. Wilson Erkrankten normal).
Differentialdiagnosen bei chronischem Verlauf
Leberzirrhosen anderer Genese, PBC, PSC, angeborene chronische Cholestasefomen (Gallengangsatresie, Alagille Syndrom).
Befunde, die gelegentlich keine eindeutige Schlussfolgerung zulassen:
- Coeruloplasmin i. S. ist meistens erniedrigt, kann aber auch bei anderen Leberzirrhosen im Stadium der Dekompensation erniedrigt sein;
- der Kupfergehalt der Leber ist erhöht, er kann aber auch bei chronischen Cholestasen, wie PBC und PSC, erhöht sein;
- der Kayser-Fleischer-Kornealring kann – vor allem in den Anfangsstadien – fehlen.
Therapie der Wilson-Krankheit
Die Behandlung des Morbus Wilson basiert auf kupferarmer Kost und Medikamenten, welche die Kupferausscheidung fördern und die Kupferaufnahme im Darm hemmen. Bei schwerem Verlauf kommt eine Lebertransplantation infrage. Neue Therapieoptionen zeichnen sich ab.
→ Auf facebook informieren wir Sie über Neues und Interessantes.
→ Verwalten Sie Ihre Laborwerte mit der Labor-App Blutwerte PRO – mit Lexikonfunktion.
Verweise
Weiteres
- Nat Rev Dis Primers. 2018;4(1):21. doi: 10.1038/s41572-018-0018-3[↩]
- Hum Genet. 2020 Aug;139(8):1065-1075[↩]
- Genet Med. 2019 May;21(5):1155-1163.[↩]
- Hepatology. 2020;71(2):722–732[↩]
- Brain. 2013 May;136(Pt 5):1476-87[↩]
- Brain. 2013 May;136(Pt 5):1476-87. doi: 10.1093/brain/awt035[↩]
- BMC Gastroenterol. 2021 Sep 1;21(1):339. doi: 10.1186/s12876-021-01911-5[↩]
- World J Hepatol. 2021 Jun 27;13(6):634-649. DOI: 10.4254/wjh.v13.i6.634 .[↩]
- Front Cell Dev Biol. 2022 May 2;10:871877. DOI: 10.3389/fcell.2022.871877[↩]
- Eur J Hum Genet. 2010 May;18(5):511-8. doi: 10.1038/ejhg.2009.187[↩]
- Therap Adv Gastroenterol. 2017;10(11):889–905. doi: 10.1177/1756283X17731520[↩]
- Pharmacol Rep. 2021 Oct;73(5):1427-1438. doi: 10.1007/s43440-021-00290-8[↩]
- J Hematol Oncol. 2024 Aug 16;17(1):68. doi: 10.1186/s13045-024-01589-8[↩]
- Parkinsonism Relat Disord. 2018 Jan 4. pii: S1353-8020(18)30007-5. doi: 10.1016/j.parkreldis.2018.01.007.[↩][↩]
- Mymensingh Med J. 2014 Jan;23(1):195-203.[↩]
- Tremor Other Hyperkinet Mov (N Y). 2023 Oct 9;13:37. doi: 10.5334/tohm.794.[↩]
- Am J Med. 1979 Aug;67(2):249-54. DOI: 10.1016/0002-9343(79)90399-1[↩]
- Osteoporos Int. 2018 Feb;29(2):315-322. DOI: 10.1007/s00198-017-4295-6[↩]
- BMJ Case Rep. 2011 Aug 11;2011:bcr0420114121. DOI: 10.1136/bcr.04.2011.4121[↩]
- Osteoporos Int. 2014 Nov;25(11):2573-80. DOI: 10.1007/s00198-014-2806-2[↩]
- Gastroenterology. 2007 Apr;132(4):1294-8[↩]
- Cochrane Database Syst Rev. 2019 Nov 19;2019(11):CD012267. doi: 10.1002/14651858.CD012267.pub2[↩][↩]
- PLoS One. 2018 Jan 11;13(1):e0190887. doi: 10.1371/journal.pone.0190887[↩]
- Ann Gen Psychiatry. 2017 Apr 4;16:19. doi: 10.1186/s12991-017-0142-6[↩]
- Therap Adv Gastroenterol. 2017 Nov;10(11):889-905. doi: 10.1177/1756283X17731520. Epub 2017 Oct 3.[↩]